
Environmental Compliance for Pharmaceutical Manufacturing in India | EHSSaral
Pharmaceutical Industry Pharma EHS Compliance Environmental Compliance India Pollution Control CPCB & SPCB Regulations Industrial Compliance EHS Management
Last updated:
|10 Feb 2026
Read time: 20 min read
A Practical, System-Level Guide for EHS & Plant Teams
Why Pharmaceutical Compliance Feels Different
Environmental compliance in pharmaceutical manufacturing does not behave like compliance in most other industries.
In many sectors, environmental non-compliance is treated as a regulatory risk - a notice, a corrective action, sometimes a penalty. In pharmaceuticals, it often becomes an existential risk. One serious observation can halt production, disrupt exports, trigger parallel scrutiny from quality teams, and place senior management directly in the line of accountability.
This difference is not accidental. It is structural.
Pharmaceutical manufacturing sits at the intersection of environmental protection, drug safety, and public health. Regulators are not only concerned about emissions or effluent in isolation; they are concerned about what those pollutants represent. Solvents, intermediates, residues, and biological materials used in pharma processes are often toxic, persistent, or bio-active. A lapse here is not seen as an industrial inconvenience - it is viewed as a potential public health event.
Batch Manufacturing Changes Everything
One of the most important reasons pharma compliance behaves differently is the nature of production.
Unlike continuous industries (cement, power, refineries) where inputs and outputs are relatively stable, most pharmaceutical units - especially API and intermediate manufacturers - operate on batch processes. This means:
Effluent characteristics can change sharply from one shift to another
Solvent consumption varies with product mix
Emission profiles fluctuate depending on reaction chemistry
Pollution loads are episodic, not averaged
From a regulatory perspective, this creates uncertainty. A clean sample taken today does not guarantee that the same system was compliant yesterday or will be compliant tomorrow. Regulators know this, which is why pharmaceutical plants are often examined with a higher degree of skepticism.
Inspections are not just about “meeting limits.” They are about process integrity - whether the systems in place can handle variability without breakdowns, bypasses, or data manipulation.
Two Regulators, One Factory
Pharmaceutical plants also carry a burden few industries do: dual regulatory accountability.
On one side are environmental regulators - CPCB and SPCBs - focused on emissions, effluent, waste, and compliance documentation. On the other side are drug regulators and quality auditors - CDSCO, USFDA, EU agencies - focused on data integrity, traceability, and process discipline.
The overlap is subtle but dangerous.
A gap in environmental documentation (for example, inconsistent reactor temperature or solvent usage logs) may trigger:
An environmental observation for non-compliance, and
A quality or data-integrity concern from a drug auditor.
This is why environmental compliance failures in pharma rarely stay “environmental.” They cascade.
Compliance Stress Comes From Complexity, Not Negligence
In many Indian pharmaceutical plants, compliance stress does not arise because teams are careless or unethical. It arises because the system is complex.
Multi-product facilities generate:
Large volumes of variable data
Multiple waste streams
Parallel registers and returns
Dozens of consent conditions with different timelines
EHS teams are expected to manage this while coordinating with production, QA, utilities, stores, and external vendors. When something slips, it is usually not due to intent - it is due to fragmentation.
Understanding this reality is the first step toward managing pharma compliance properly.
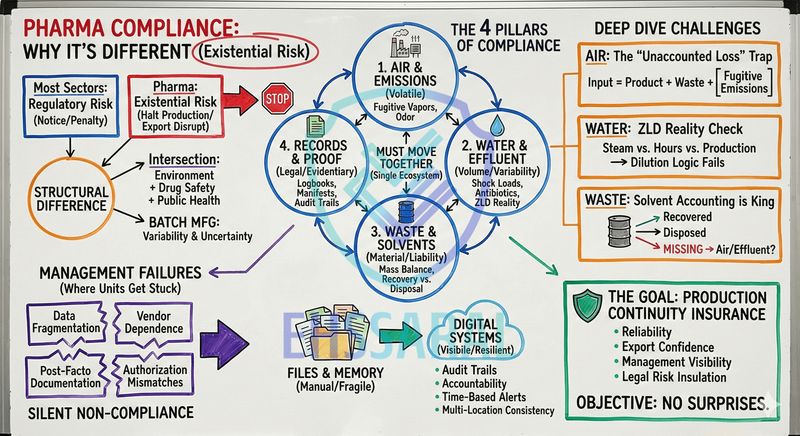
The Four Pillars of Environmental Compliance in Pharma
To make sense of pharmaceutical compliance, it helps to move away from rule-by-rule thinking and adopt a system framework.
In practice, environmental compliance in pharma rests on four interconnected pillars. Treating them separately creates gaps. Managing them together creates stability.
1. Air & Emissions - The Volatile Risk
This pillar goes far beyond the boiler stack.
In pharmaceutical plants, air compliance includes:
Reactor vents
Scrubbers
Solvent storage and handling
Fugitive emissions from equipment
Odour control
Many pharma units appear compliant on paper because their stacks meet limits. Yet enforcement actions often arise from invisible emissions - vapours, leaks, or odours that do not show up neatly in reports.
In practice, odour is often the first whistleblower.
2. Water & Effluent - The Volume and Variability Risk
Pharma effluent is not just high in volume; it is high in variability.
COD, TDS, pH, and toxicity can change dramatically based on:
Product mix
Cleaning cycles
Batch sequencing
Managing this pillar is less about treatment capacity and more about segregation, equalization, and shock-load control. Systems that work well for steady industries often struggle here.
3. Waste & Solvents - The Material and Liability Risk
This is the most scrutinized pillar in pharmaceutical plants.
Spent solvents, mother liquor, API residues, contaminated packaging, and process sludge are all high-liability materials. Regulators focus heavily on:
Recovery ratios
Material balance
Authorized handling and disposal
Any mismatch between purchase, recovery, disposal, and production raises immediate questions.
4. Records, Returns & Proof - The Legal Risk
The first three pillars are physical. This pillar is evidentiary.
It includes:
Consent to Operate conditions
Logbooks and daily records
Manifests and waste returns
Annual submissions
In pharma, paper reality must match physical reality. If the system works but records don’t reflect it, the plant is still considered non-compliant.
Why These Pillars Must Move Together
These four pillars do not operate independently.
You cannot:
Fix effluent issues without addressing solvent recovery
Prove air compliance without reliable logbooks
Defend waste handling without consistent material balance
They form a single ecosystem. Weakness in one pillar destabilizes the others.
This is where confusion usually starts - teams try to “fix” one area in isolation, only to discover the problem reappearing somewhere else.
Air Emissions & Process Vents in Pharmaceutical Plants
Air compliance in pharmaceutical plants is often misunderstood because it looks simple on paper.
A boiler stack meets limits. DG sets are tested. Monitoring reports are available. On the surface, everything appears under control. Yet many pharma units face environmental actions not because of stack failures, but because of fugitive and process emissions.
This is where pharma differs sharply from most other industries.
Reactor Vents and Scrubber Reality
In API and intermediate manufacturing, reactors are the heart of operations - and also the heart of air emission risk.
During reactions, especially those involving solvents, acids, or halogenated compounds, vapours are released through:
Reactor vents
Condensers
Pressure relief systems
Most plants rely on scrubbers to manage these emissions. The issue is not the presence of a scrubber, but its design logic and operational discipline.
Common gaps seen in practice:
Scrubber media selected for average conditions, not worst-case batches
Infrequent replacement or regeneration of scrubbing solution
No calculation of scrubbing efficiency against peak vapour loads
Scrubbers running mechanically, but not chemically effective
From an inspector’s standpoint, a scrubber that exists but is not demonstrably effective is equivalent to no scrubber at all.
Solvent Vapours and the “Unaccounted Loss” Trap
One of the most common audit traps in pharmaceutical plants is solvent mass balance.
Inspectors increasingly apply a simple but powerful logic:
Solvent Purchased = Solvent in Product + Solvent Recovered + Solvent Disposed
If this equation does not balance, the remainder is assumed to have escaped into the environment - primarily through air.
For example:
1,000 litres purchased
700 litres recovered
200 litres disposed
100 litres unaccounted
That 100 litres becomes a fugitive emission assumption, even if no visible leak is detected.
This is why air compliance in pharma cannot be evaluated independently of solvent records, recovery logs, and waste manifests. The system is judged holistically.
Odour: The Hidden Compliance Trigger
Legally, odour is categorized as a nuisance. Practically, it is one of the most powerful enforcement triggers.
In industrial estates across India, a large proportion of unplanned inspections begin with:
A resident complaint
A neighbouring unit reporting chemical smell
A security guard flagging strong odour at the gate
Once an inspection begins with odour, the bias is already negative. Inspectors are no longer just verifying compliance - they are looking for confirmation of suspicion.
Controlling odour is not about aesthetics. It is about inspection risk management.
Plants that proactively manage odour through:
Proper vent condensation
Adequate scrubbing
Solvent handling discipline
often experience fewer surprise inspections, even if their operations are otherwise intensive.
Effluent, ZLD, and the Pharma ETP Reality
If air emissions represent the volatile risk in pharma, effluent represents the systemic risk.
Pharmaceutical effluent is challenging not only because of its strength, but because of its unpredictability.
COD, TDS, and Shock Loads
In multi-product pharmaceutical facilities, effluent characteristics can swing dramatically:
One batch may generate COD of 5,000 mg/L
Another may exceed 50,000 mg/L
Cleaning cycles add sudden dilution or chemical spikes
Conventional biological treatment systems are not designed to handle such shock loads gracefully. Without aggressive equalization and segregation, systems oscillate between overloading and underutilization.
This is why many pharma ETP failures are not equipment failures - they are control failures.
Equalization Is Not a Tank, It’s a Strategy
Many plants treat the equalization tank as a passive buffer. In pharma, it must be actively managed.
Effective equalization requires:
Controlled feed from different streams
Continuous monitoring of influent strength
Operational coordination with production schedules
When equalization is weak, downstream biological systems suffer. Sludge bulking, foaming, and crashes follow - and once the system destabilizes, compliance becomes reactive.
Antibiotic Residues: A New Compliance Frontier
Recent regulatory attention has shifted toward antibiotic residues in treated effluent.
The concern is not just water pollution, but the development of antimicrobial resistance (AMR). As a result, regulators are increasingly questioning:
Whether antibiotic-containing streams are segregated
Whether pre-treatment or kill tanks are used
Whether advanced oxidation processes (AOP) are required
This marks a shift from “end-of-pipe treatment” to source-level control. Pharma plants that do not rethink their effluent segregation strategy may find their existing ETP designs questioned, even if historical compliance exists.
ZLD: Claims vs Operational Reality
Zero Liquid Discharge (ZLD) is effectively mandatory for most bulk drug and API units in India. However, enforcement is no longer limited to checking whether a Multiple Effect Evaporator (MEE) exists.
Inspectors now examine:
Steam consumption patterns
Condensate quality
MEE operating hours vs production levels
A common red flag arises when:
Production is high
Effluent generation is high
MEE steam usage is low
The inference is straightforward: bypass or dilution.
In pharma, dilution logic fails because regulators understand the pollution load per kg of product. Numbers that don’t align raise immediate suspicion.
Hazardous Waste, Spent Solvents & Mother Liquor
Among all compliance areas, this is where pharmaceutical plants face the highest scrutiny - and the highest liability.
Why Solvent Accounting Is King
Solvents are the bloodstream of pharmaceutical manufacturing. They are also one of the most valuable and most regulated inputs.
Regulators expect:
High recovery efficiency
Clear differentiation between recoverable and disposable streams
Transparent documentation
If the Consent to Operate states a 95% recovery rate but records show only 80%, the gap is not seen as inefficiency - it is seen as misrepresentation.
Where did the remaining solvent go?
Into the air?
Into the effluent?
Into unauthorized sale channels?
Each possibility has regulatory consequences.
Recovery vs Disposal: A Critical Distinction
A recurring issue in inspections is the misclassification of waste.
Spent solvent implies recoverability
Hazardous waste implies disposal
Mother liquor sits in a grey zone and is frequently misused. Inspectors are alert to situations where mother liquor is shown as spent solvent to justify sale to recyclers who are not authorized to handle it.
This is not a documentation issue - it is a classification integrity issue.
Manifest Patterns Inspectors Watch Closely
Hazardous waste manifests are legal transport documents. Patterns matter more than individual entries.
Common red flags include:
Missing or skipped manifest serial numbers
Mismatch between waste generated and waste dispatched
Transporters lacking authorization for specific waste categories
Once inconsistencies are detected, inspectors often widen the scope to past returns and storage practices.
Storage Area: The First Walk
In many inspections, the hazardous waste storage area is the first physical location visited.
Inspectors typically check:
Segregation of incompatible waste
Date labeling on containers (90-day storage rule)
Spill containment and secondary bunding
Housekeeping and access control
A poorly managed storage area sets the tone for the entire inspection.
Compliance Differences Across Pharmaceutical Manufacturing Types
One of the most common mistakes in pharmaceutical compliance discussions is treating the industry as a single category. In reality, regulatory expectations vary significantly based on the nature of pharmaceutical operations.
Inspectors, whether formally or informally, apply different mental benchmarks depending on what kind of pharma unit they are evaluating. Understanding this distinction helps EHS teams anticipate scrutiny instead of reacting to it.
API Manufacturing (Bulk Drugs)
Risk Profile: Critical
Regulatory Lens: Pollution load per kg of product
API units attract the highest level of environmental scrutiny because they generate:
High-strength effluent
Large solvent volumes
Toxic and persistent residues
From a compliance standpoint, API manufacturing is evaluated less on absolute limits and more on process credibility.
Inspectors focus heavily on:
Solvent recovery efficiency
ZLD system performance
Material balance across batches
Incineration and TSDF routing
Even small inconsistencies carry weight because regulators assume that minor deviations at API scale translate into major environmental impact.
A recurring observation in API units is that equipment capacity exists, but operational discipline lags behind production intensity. This gap is where most enforcement actions originate.
Formulation Units (Tablets, Capsules, Syrups)
Risk Profile: Moderate
Regulatory Lens: Documentation discipline
Formulation plants typically generate lower pollution loads compared to API units. However, this does not translate to relaxed enforcement.
In practice, formulation units face:
Equal or higher documentation scrutiny
Greater focus on housekeeping and segregation
Attention to wash water, floor cleaning effluent, and packaging waste
Because pollution loads are relatively low, inspectors expect near-perfect documentation. Missing entries, delayed logbooks, or expired authorizations are less likely to be overlooked.
A common misconception is that formulation plants are “low-risk.” In reality, they are low-tolerance units - documentation errors stand out more sharply.
Biotech, Sterile & Injectable Manufacturing
Risk Profile: High
Regulatory Lens: Bio-safety and containment
Biotech and sterile facilities introduce an additional layer of concern: biological risk.
Regulators focus on:
Inactivation of biological waste
Sterilization procedures
Handling of live cultures and contaminated sharps
Disposal of single-use plastics
Here, environmental compliance overlaps strongly with biosafety. Even minor lapses raise alarms because the consequences are perceived as uncontrolled biological release, not just pollution.
Systems that work for chemical waste often fail here because biological risks demand absolute containment, not dilution or recovery.
R&D Facilities and Pilot Plants
Risk Profile: High (despite low volumes)
Regulatory Lens: Unpredictability
R&D and pilot-scale operations are often underestimated internally because they generate small quantities. Regulators, however, see them differently.
Key challenges include:
High variety of chemicals
Frequent process changes
Temporary setups that become permanent
Poor tracking of small but hazardous quantities
Many R&D units operate under simplified consent conditions, but this does not exempt them from hazardous waste tracking, storage norms, or authorized disposal.
When incidents occur in R&D facilities, enforcement tends to be strict because regulators view them as poorly controlled risk zones.
What Inspectors Actually Check in Pharma Units
Inspection outcomes in pharmaceutical plants are often decided before formal sampling begins.
Experienced inspectors rely on signals, not just checklists. These signals indicate whether the plant is under control or merely compliant on paper.
Material Balance Logic
Inspectors often start with inputs, not outputs.
They review:
Raw material purchase records
Solvent invoices
Production batch logs
Their underlying equation is simple:
Input = Product + Waste + Emissions
If this balance does not make intuitive sense, deeper scrutiny follows. Sampling becomes secondary to investigation.
Logbook Continuity
Logbooks are evaluated for behavior, not just values.
An experienced inspector knows what real-time data looks like. It fluctuates. It has minor corrections with initials. Different shifts write differently. The pH at 2 PM doesn’t round to exactly 7.0 for three days straight. When logbooks look too “clean,” they raise suspicion - not confidence.
What typically raises flags:
- Identical handwriting across multiple shifts (suggesting one person filled everything later)
Overwriting without initials, dates, or a clear correction note
Data entries recorded during known equipment shutdowns or maintenance windows
“Perfect” readings that never show normal process variation
A particularly telling cross-check is energy consumption versus logged operating hours. If your ETP blower or pump consumed power consistent with 4–6 hours of operation, but the logbook claims it ran 24 hours, the story doesn’t align. In pharma plants, these small inconsistencies often expand into broader questions: “If the logs are reconstructed, what else is reconstructed?”
Effluent Segregation on the Ground
Paper flow diagrams are rarely trusted without physical verification.
Inspectors trace:
Pipeline routes
Valve positions
Bypass arrangements
A hidden or undocumented bypass line is one of the fastest ways to escalate an inspection.
Smell, Noise, and First Impressions
Human senses matter.
If inspectors:
Smell solvent at the gate
Hear abnormal noise from utilities
Observe poor housekeeping in the first few minutes
the inspection tone shifts immediately. From that point, the plant is inspected with assumed non-compliance until proven otherwise.
Where Pharmaceutical Units Usually Get Stuck
Most compliance failures in pharma plants are not technical failures. They are management failures.
Data Fragmentation
Information is scattered:
EHS holds the consent
Production holds solvent logs
Stores hold manifests
Utilities hold energy data
When an inspector asks for a consolidated picture, teams scramble. Delays and inconsistencies follow.
Vendor Dependence
Many plants outsource:
ETP operation
Return filing
Manifest handling
If the vendor delays or cuts corners, the liability still rests with the occupier. Vendor dependence without internal oversight is one of the most common risk amplifiers.
Post-Facto Documentation
Filling logbooks weekly instead of shift-wise leads to:
Memory-based entries
Inconsistencies
Data that “looks written” rather than generated
Inspectors detect this quickly.
Authorization Mismatches
Product mix changes are frequent in pharma. Compliance updates are not.
Producing a new molecule without updating:
Consent conditions
Hazardous waste authorization
Solvent recovery declarations
creates silent non-compliance that surfaces only during audits or renewals.
Why Pharmaceutical Compliance Is Shifting from Files to Systems
For many years, pharmaceutical environmental compliance in India survived on a combination of paper files, Excel trackers, and human memory. That model is now reaching its natural limit.
This is not because regulators suddenly prefer technology. It is because the complexity of pharma operations has crossed the threshold where manual control becomes unreliable.
Audit Trails Are No Longer Optional
In earlier years, inspectors were satisfied with the presence of records. Today, they increasingly ask:
Who entered this data?
When was it recorded?
Was it recorded in real time or reconstructed later?
Paper logbooks cannot answer these questions convincingly. Digital systems can.
An audit trail is no longer just a “nice to have.” It has become a proxy for intent. When data is time-stamped, role-tagged, and sequential, regulators infer discipline. When data looks reconstructed, they infer risk.
Accountability Must Be Visible
In pharma plants, compliance tasks are distributed:
A shift in-charge checks pH
A utility operator monitors the ETP
A store executive handles waste dispatch
An EHS officer consolidates returns
On paper, accountability is vague. In practice, it often collapses during inspections.
System-based compliance introduces clarity:
Tasks are assigned
Deadlines are visible
Missed actions are recorded
This visibility changes behavior. When responsibility is explicit, compliance becomes routine instead of reactive.
Time-Based Risk Is the Real Enemy
Most pharma compliance failures are not dramatic violations. They are missed timelines.
Examples include:
Consent renewals overlooked until expiry
Bank guarantees lapsing silently
Annual returns filed late
OCEMS calibration deadlines missed
Example: The Silent Lapse
A formulation unit in Gujarat operated smoothly for years. Their hazardous waste authorization expired in March. The renewal application was filed in April - a one-month delay.
Operationally, nothing changed. The plant continued generating waste, storing it, and dispatching it to their usual TSDF. From the factory’s point of view, everything was “normal.”
In June, during a routine inspection, the lapsed authorization was noticed. The consequence was not a small warning:
- Waste dispatched during the lapse period was flagged as “unauthorized transportation”
The TSDF’s acceptance trail was questioned
Past manifests were reviewed in detail
A show-cause notice was issued, consuming months of response and follow-up
The operational impact was zero. The legal exposure was significant.
This is time-based risk. A one-month documentation gap can become a six-month compliance recovery process. Paper calendars don’t prevent this. Systems do.
Paper files do not warn you. Systems do.
Time-based risk is invisible until it becomes irreversible. This is why regulators increasingly emphasize timelines over explanations.
Multi-Location Consistency Is Becoming Critical
Many pharmaceutical companies operate multiple plants:
Different states
Different SPCBs
Different local practices
Without centralized visibility, compliance quality varies by site. This inconsistency is increasingly unacceptable to regulators and corporate leadership alike.
Systems do not replace people, but they standardize expectations. What is acceptable at one site becomes the baseline for all sites.
Compliance as Production Continuity Insurance
One of the most damaging misconceptions in pharmaceutical manufacturing is treating environmental compliance as a cost center.
From a practical standpoint, compliance functions more like insurance.
Reliability
A plant that operates within well-understood compliance boundaries is far less likely to face:
Sudden closures
Emergency inspections
Production stoppages
The cost of one forced shutdown often exceeds years of compliance investment.
Export Confidence
Global customers do not view environmental compliance as a local issue.
USFDA and EU auditors increasingly review:
Waste handling logic
Effluent treatment practices
Data integrity in environmental records
Strong environmental compliance becomes a commercial differentiator, not just a regulatory necessity.
Management Visibility
Senior management rarely wants to know:
How many logbooks exist
They want to know:
Is the plant compliant today?
Where are the risks this month?
A system-level view enables informed decisions instead of last-minute firefighting.
Legal Risk Insulation
Environmental non-compliance increasingly carries personal liability for Directors, Occupiers, and Factory Managers.
Consistent records, clear task ownership, and demonstrable control are not just operational safeguards - they are legal shields.
Final Perspective
Environmental compliance in pharmaceutical manufacturing is not about ticking boxes. It is about maintaining control over complex, variable systems.
Plants that understand this treat compliance as a daily operational function, not an annual ritual. Over time, these plants face fewer inspections, smoother renewals, and stronger confidence from regulators and customers alike.
Good compliance does not eliminate risk.
It prevents surprises.
That is the real objective.
Frequently Asked Questions
Is ZLD mandatory for all pharmaceutical units?
Not for all units uniformly. However, for bulk drug and API manufacturing, ZLD has effectively become the default expectation in most states due to the risk of groundwater contamination. Formulation units may be permitted partial discharge depending on location and effluent characteristics, but scrutiny remains high.
Why are pharmaceutical inspections stricter than other industries?
Pharma pollutants are often toxic, persistent, or bio-active. Regulators also know that treatment costs are high, creating a temptation to bypass systems. Combined with public health sensitivity, this leads to stricter enforcement.
How are solvents evaluated during audits?
Inspectors use a mass balance approach:
Solvent purchased
Solvent consumed
Solvent recovered
Solvent disposed
Any unexplained gap is assumed to be environmental loss and is questioned accordingly.
We recovered 85% solvent but our consent says 95%. Are we non-compliant?
Technically, yes - but the real question inspectors ask is “why?” If 85% is your best achievable recovery for a specific chemistry, the solution is not to make the records look like 95%. The solution is to document the limitation, generate a justified recovery basis, and update your consent conditions accordingly.
Showing 95% on paper when actual recovery is 85% creates a “ghost gap” that will eventually surface in your material balance - and then the assumption becomes: the missing fraction went into air or effluent. That is a much harder conversation than a justified consent update.
Our MEE runs only 6 hours/day but we claim ZLD. Is this a problem?
It depends on your effluent generation and your MEE’s rated evaporation capacity. If you generate 40–60 kL/day of concentrated stream but the MEE’s practical throughput requires 12+ hours to process that volume, the math doesn’t work.
Inspectors increasingly cross-check three things together: steam consumption, operating hours, and claimed evaporation rates. If these don’t align, bypass or dilution is assumed - even if the equipment exists and is functional.
Can we store spent solvent and mother liquor together?
No. Even if both are liquid organic streams, they have different recovery potential and often different disposal pathways. Mixing them creates classification confusion and makes it impossible to demonstrate segregation during inspections.
This is a common violation that feels minor internally, but it signals poor hazardous waste discipline externally - and once that signal is received, the inspection scope typically widens.
Are R&D laboratories treated differently?
They may have simplified consent procedures, but they are not exempt from hazardous waste rules. Disposal through authorized channels and proper documentation are still mandatory.
What typically delays consent renewals for pharma units?
Common reasons include:
Discrepancies in hazardous waste annual returns
Changes in product mix not reflected in consent conditions
Non-compliance with previous consent stipulations
Pending OCEMS or monitoring-related observations

Harshal T Gajare
Founder, EHSSaral
Founder - EHSSaral| Partner - Perfect Pollucon | ISO 14001 Lead Auditor | Second-generation environmental professional simplifying EHS compliance for Indian manufacturers through practical, tech-enabled guidance.
Related Blogs

Environmental Compliance in India: Beginner Roadmap | EHSShala

Why BMW Inspections Feel Stricter Today (India) | EHSSaral

New Labour Codes in India Effective Nov25: Mandatory Appointment Letters & Health Checks | EHSSaral

Why Safety Culture Fails in Indian SMEs: People & Compliance Challenges | EHSSaral Research

Late Filing Penalty for Form 4 SPCB: What Happens If You Miss the 30 June Deadline?
Environmental Monitoring Guide for Indian Factories | EHSShala
How to Fill ESG & Sustainability Vendor Forms for SMEs in India

EHSShala Foundations - Core Environmental Basics for EHS Officers
